La gemcitabina è un analogo nucleosidico, ampiamente utilizzato da solo o in combinazione, per il trattamento di più tumori. La gemcitabina è generalmente preferita nei pazienti anziani o fragili a causa del profilo di tossicità inferiore rispetto ad altri farmaci antitumorali. Tuttavia, la gemcitabina può anche essere associata a reazioni avverse cardiovascolari (CV-ADR). Infatti, oltre alla mielosoppressione, sono emerse molte altre ADR da quando la gemcitabina è stata approvata dalla Food and Drug Administration (FDA), inclusa la microangiopatia trombotica, polmonite interstiziale e sindrome da perdita capillare (CLS).
Un articolo pubblicato recentemente su Pharmaceuticals, fornisce una revisione della letteratura e racconta uno studio di farmacovigilanza osservazionale, retrospettivo, effettuato attraverso Vigibase.
Revisione in letteratura
È stata effettuata una ricerca dei casi di cardiotossicità associati alla gemcitabina, pubblicati su MEDLINE fino al 30 maggio 2019.
Gli autori hanno individuato 23 casi di reazioni avverse cardiovascolari associate a gemcitabina. Di questi: 4 erano casi di infarto del miocardio, 10 casi di insufficienza cardiaca, 6 casi di aritmie sopraventricolari, 8 casi di disturbi associati al pericardio.
I pazienti avevano assunto gemcitabina per il trattamento di: cancro al pancreas (12 casi, 52%), cancro al polmone (5 casi, 22%), linfoma (5 casi, 22%).
La gemcitabina è stata sospesa definitivamente in 16/23 casi (70%) e in 7 è stata ripresa. La ricomparsa dei sintomi in seguito alla riassunzione del farmaco (rechallenge positivo) si è verificata in 4/7 casi (57%).
In questa revisione sono stati esaminati anche i trials clinici sulla gemcitabina. Di 106 studi (per un totale di 14015 pazienti coinvolti), in 17 (pazienti totali 2386) sono state segnalati 33 casi di reazioni avverse cardiovascolari. Da questi dati, sono stati stimati dei tassi di incidenza: 0,24% (33/14015) per tutti i pazienti che hanno ricevuto gemcitabina nei trials clinici e 1,38% (33/2386) per i pazienti che hanno partecipato agli studi nei quali sono state segnalate le ADR cardiovascolari.
Delle 33 reazioni cardiovascolari segnalate per gemcitabina, 27 erano gravi e hanno incluso: 8 casi di infarto del miocardio, 2 casi di versamento pericardico, 7 casi di insufficienza cardiaca e 1 caso di aritmia.
Studio di farmacovigilanza
Tramite VigiBase, il database globale dell’Organizzazione Mondiale della Sanità dei rapporti sulla sicurezza dei singoli casi, sono state confrontate le segnalazioni di ADR cardiovascolari associate alla gemcitabina rispetto alla totalità delle segnalazioni presenti nell’intero database fino al 1 aprile 2019.
Questo studio ha permesso di caratterizzare meglio le reazioni avverse cardiovascolari associate a gemcitabina, in particolare le caratteristiche cliniche tra cui il tempo di insorgenza e la gravità di circa 1000 segnalazioni.
Al 1 aprile 2019 il numero totale di ADR in VigiBase era 18.908.940, mentre quelle da gemcitabina erano 46.898.
La gemcitabina è stata associata a segnalazioni più elevate di ischemia miocardica (IM, n: 119), malattie pericardiche (n: 164), aritmie sopraventricolari (SVA, n: 308) e insufficienza cardiaca (HF, n: 484) rispetto alla totalità delle segnalazioni presenti nell’intero database con IC 025 compreso tra 0,40 e 2,81.
Le ADR cardiovascolari segnalate sono state associate a morte fino al 17% dei casi.
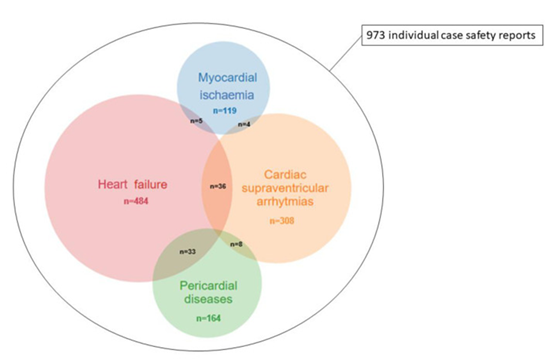
Il trattamento con gemcitabina è significativamente associato a reazioni avverse cardiovascolari potenzialmente letali, inclusi infarto del miocardio, malattie pericardiche, aritmie sopraventricolari, e insufficienza cardiaca. Questi eventi devono essere considerati nella cura del paziente e nella progettazione degli studi clinici.
Leggi l’articolo intero qui.