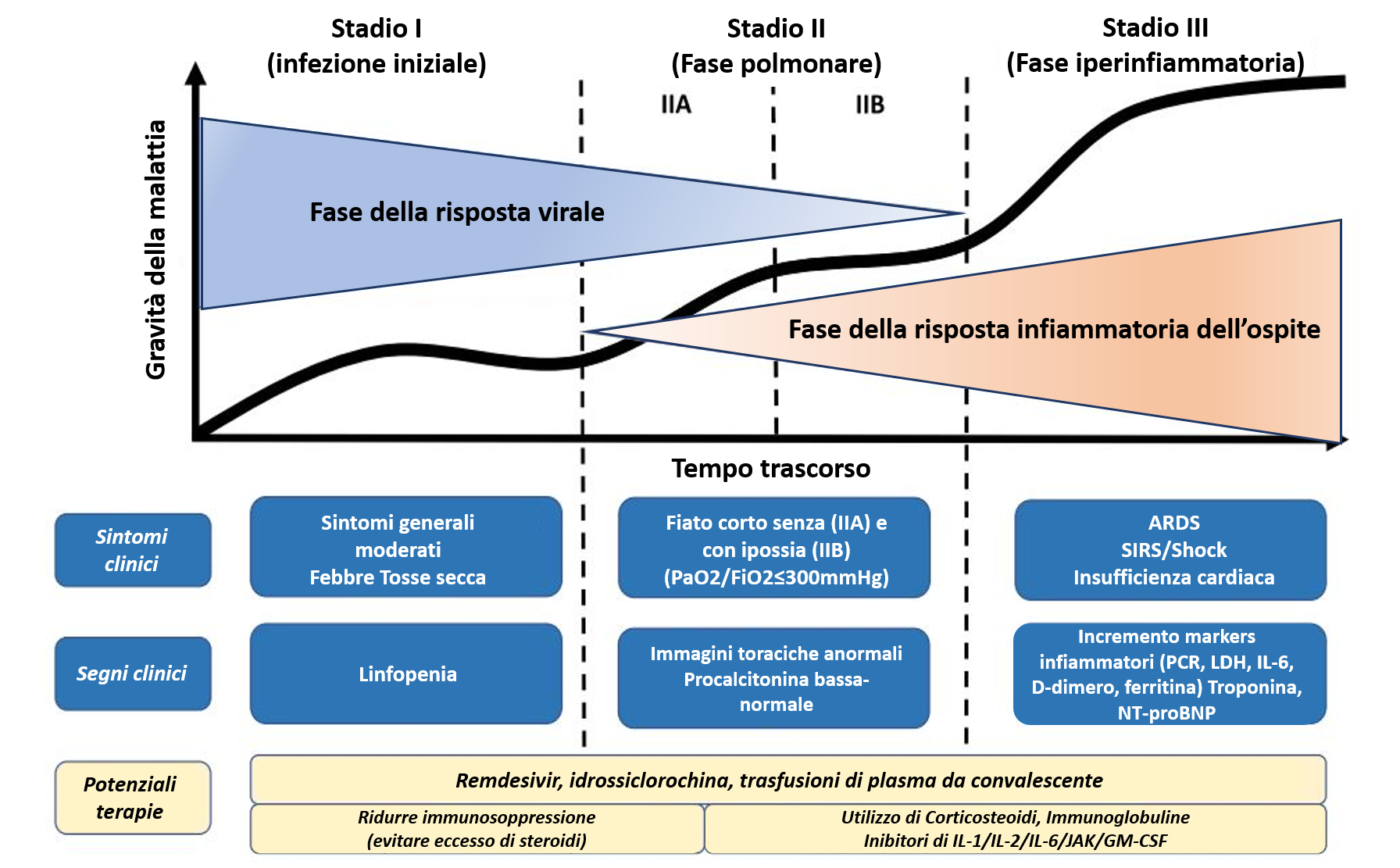
L’Agenzia Italiana del Farmaco ha pubblicato il primo Rapporto di farmacovigilanza sui vaccini COVID-19, che avrà cadenza mensile. I dati raccolti e analizzati riguardano le segnalazioni di sospetta reazione avversa registrate nella Rete Nazionale di Farmacovigilanza tra il 27 dicembre 2020 e il 26 gennaio 2021 per i vaccini in uso nella campagna vaccinale in corso: Comirnaty di Pfizer/BioNTech (autorizzato dal 22/12/2020 e utilizzato dal 27/12/2020) e COVID-19 Vaccino Moderna (autorizzato dal 07/01/2021 e utilizzato dal 14/01/2021).
Le segnalazioni riguardano soprattutto la prima dose del vaccino Comirnaty (99%), che è stato il più utilizzato e solo in minor misura il vaccino Moderna (1%).
Nel periodo considerato sono pervenute 7.337 segnalazioni su un totale di 1.564.090 dosi somministrate (tasso di segnalazione di 469 ogni 100.000 dosi), di cui il 92,4% sono riferite a eventi non gravi, come dolore in sede di iniezione, febbre, astenia/stanchezza, dolori muscolari. Con Comirnaty sono state osservate anche cefalea, parestesie, vertigini, sonnolenza e disturbi del gusto mentre con il vaccino Moderna, nausea e dolori addominali.
Meno frequenti sono le altre reazioni locali e i dolori articolari diffusi. Come atteso, la febbre è stata segnalata con maggior frequenza dopo la seconda dose rispetto alla prima.
Gli eventi segnalati insorgono prevalentemente lo stesso giorno della vaccinazione o il giorno successivo (85% dei casi).
Del 7,6% di segnalazioni classificate come “gravi”, per le quali è in corso la valutazione del nesso causale con i vaccini, tre su quattro non hanno richiesto intervento specifico in ambito ospedaliero.
Nel periodo sono stati segnalati anche 13 decessi avvenuti nelle ore successive alla vaccinazione che, nelle segnalazioni più dettagliate e complete di dati, non sono risultati correlati alla vaccinazione e sono in larga parte attribuibili alle condizioni di base della persona vaccinata.
Le analisi condotte sui dati fin qui acquisiti confermano quindi un buon profilo di sicurezza di questi due vaccini a mRNA. L’ampio numero di segnalazioni non implica che siano emerse criticità inattese, ma è indice dell’elevata capacità del sistema di farmacovigilanza nel monitorare la sicurezza.
Qui è possibile scaricare il primo rapporto sulla sorveglianza dei vaccini contro COVID-19.
Qui il comunicato stampa dal sito AIFA.